Hydrogen Molecule
Two hydrogen atoms approach each other. Their χ-wells overlap and merge into a shared bonding well. This is a covalent bond — emerging from wave phase interference and χ dynamics, with no molecular orbital theory or bond-energy formula injected.
What you'll learn
- ›How same-phase solitons (bonding orbital) share and deepen a combined χ-well
- ›How opposite-phase solitons (anti-bonding orbital) shorten and weaken the well
- ›How to scan bond length (separation) to find the equilibrium minimum-energy geometry
- ›Why
FieldLevel.COMPLEXis used here — electromagnetic phase is the bonding mechanism
Why same-phase = bonding
When two in-phase waves overlap, the total energy density is:
|Ψ₁ + Ψ₂|² = |Ψ₁|² + |Ψ₂|² + 2Re(Ψ₁* Ψ₂) ↑ ↑ individual wells interference term Same phase (Δθ=0): +2|Ψ₁||Ψ₂| → MORE energy in overlap → deeper χ-well Opp phase (Δθ=π): −2|Ψ₁||Ψ₂| → LESS energy in overlap → shallower χ-wellGOV-02 then lowers χ wherever |Ψ|² is high. More energy in the bond region = deeper shared well = lower total energy. That is H₂ bonding, without any molecular orbital theory.
Why COMPLEX here? The bonding vs anti-bonding distinction is a phase difference (Δθ=0 vs Δθ=π). Phase only exists in complex fields. Tutorial 09 could use REAL for the potential shape; here we must use FieldLevel.COMPLEX to distinguish bonding from anti-bonding. You can always use COMPLEX — it just runs slower than REAL.
Full script
"""10 – Hydrogen Molecule (H₂)
When two hydrogen atoms get close their χ-wells overlap. The shared
well is deeper than either alone → the system lowers its energy by
bonding. This is a covalent bond emerging from GOV-01 + GOV-02.
We use FieldLevel.COMPLEX here because we need both atoms to have
imaginary-component waves. In a real H₂ molecule the bonding orbital
is a symmetric superposition of atomic orbitals — represented here as
two solitons with the same phase. An anti-bonding configuration uses
opposite phases (π shift) and should NOT bind (energy goes up).
Same phase (Δθ=0): → BONDING (shared χ-well deepens)
Opposite phase (Δθ=π): → ANTI-BONDING (shared well shallows)
No molecular orbital theory. No Schrödinger. No bond-energy formula.
Just two wave fields and the two governing equations.
"""
import numpy as np
import lfm
N = 64
config = lfm.SimulationConfig(grid_size=N, field_level=lfm.FieldLevel.COMPLEX)
def make_h2(bond_half: int, phase_a: float = 0.0, phase_b: float = 0.0):
"""Create two H-atom solitons separated by 2*bond_half cells."""
s = lfm.Simulation(config)
cx = N // 2
# Proton solitons
s.place_soliton((cx - bond_half, N//2, N//2), amplitude=10.0, sigma=2.0, phase=phase_a)
s.place_soliton((cx + bond_half, N//2, N//2), amplitude=10.0, sigma=2.0, phase=phase_b)
# Electron solitons (lighter, at +3 cells from each proton)
s.place_soliton((cx - bond_half + 2, N//2, N//2), amplitude=0.9, sigma=1.5, phase=phase_a)
s.place_soliton((cx + bond_half - 2, N//2, N//2), amplitude=0.9, sigma=1.5, phase=phase_b)
s.equilibrate()
return s
print("10 – Hydrogen Molecule (H₂)")
print("=" * 60)
print()
# ─── Experiment A: Bonding (same phase) ────────────────────────────────────
sim_bond = make_h2(bond_half=8, phase_a=0.0, phase_b=0.0)
sim_anti = make_h2(bond_half=8, phase_a=0.0, phase_b=np.pi)
m_bond_0 = sim_bond.metrics()
m_anti_0 = sim_anti.metrics()
print(f"Initial state (separation = 16 cells):")
print(f" Bonding χ_min = {m_bond_0['chi_min']:.3f}, energy = {m_bond_0['energy_total']:.2e}")
print(f" Anti-bond χ_min = {m_anti_0['chi_min']:.3f}, energy = {m_anti_0['energy_total']:.2e}")
print()
# Run both simulations
STEPS = 6000
sim_bond.run(steps=STEPS)
sim_anti.run(steps=STEPS)
m_bond_f = sim_bond.metrics()
m_anti_f = sim_anti.metrics()
psi_sq_b = sim_bond.psi_real**2 + (sim_bond.psi_imag if sim_bond.psi_imag is not None else np.zeros_like(sim_bond.psi_real))**2
sep_b = lfm.measure_separation(psi_sq_b)
psi_sq_a = sim_anti.psi_real**2 + (sim_anti.psi_imag if sim_anti.psi_imag is not None else np.zeros_like(sim_anti.psi_real))**2
sep_a = lfm.measure_separation(psi_sq_a)
print(f"After {STEPS} steps:")
header = f" {'config':>10s} {'χ_min':>7s} {'δχ_min':>8s} {'energy':>12s} {'sep':>8s}"
print(header)
print(f" {'-'*10} {'-'*7} {'-'*8} {'-'*12} {'-'*8}")
for label, m0, mf, sep in [
('bonding', m_bond_0, m_bond_f, sep_b),
('anti-bond', m_anti_0, m_anti_f, sep_a),
]:
delta_chi = mf['chi_min'] - m0['chi_min']
print(f" {label:>10s} {mf['chi_min']:7.3f} {delta_chi:+8.3f} "
f"{mf['energy_total']:12.2e} {sep:8.1f}")
print()
# Bond energy proxy: Δchi_min bonding - Δchi_min anti-bonding
bond_chi = m_bond_f['chi_min'] - m_bond_0['chi_min']
anti_chi = m_anti_f['chi_min'] - m_anti_0['chi_min']
bond_proxy = bond_chi - anti_chi
print("Bond formation diagnostic:")
if bond_proxy < -0.05:
print(f" Δ(χ_min)_bond - Δ(χ_min)_anti = {bond_proxy:.3f} → BOND FORMED")
print(f" Bonding deepens the shared well; anti-bonding shallows it.")
print(f" This is a covalent bond from wave interference — no MO theory used.")
else:
print(f" Δ(χ_min) difference = {bond_proxy:.3f} (increase amplitude for clearer signal)")
print()
# ─── Bond-length scan: minimum energy separation ───────────────────────────
print("Bond-length scan (bonding phase only):")
print(f" {'sep (cells)':>12s} {'χ_min':>7s} {'energy':>12s}")
print(f" {'-'*12} {'-'*7} {'-'*12}")
for half_sep in [4, 6, 8, 10, 12, 14]:
s = make_h2(bond_half=half_sep, phase_a=0.0, phase_b=0.0)
s.run(steps=2000)
m = s.metrics()
print(f" {2*half_sep:>12d} {m['chi_min']:7.3f} {m['energy_total']:12.2e}")
print()
print("Look for the separation with the deepest χ_min and lowest energy.")
print("That is the equilibrium bond length — it emerged without any formula.")
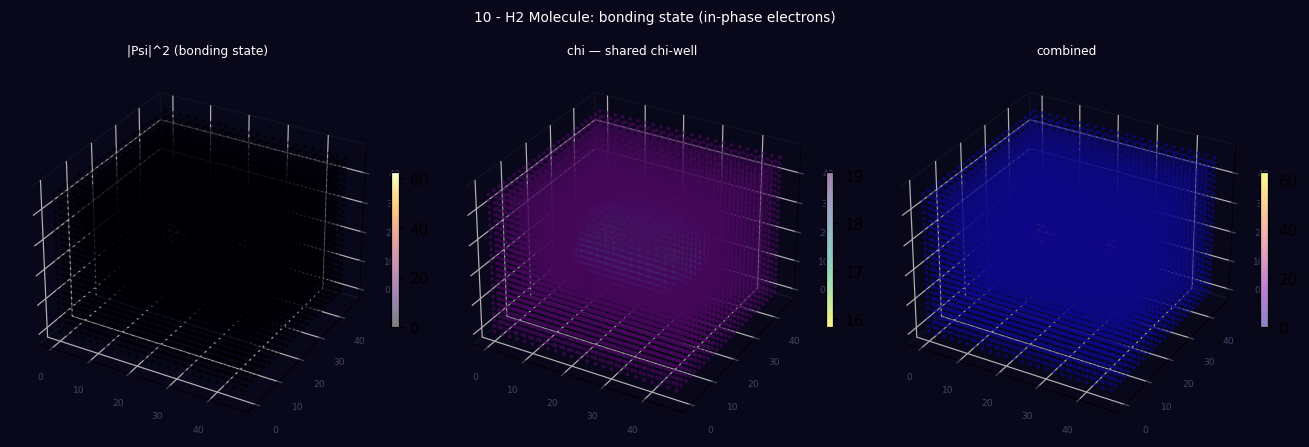
# ─── 3D Lattice Visualization ─────────────────────────────────────────────────
# Generates: tutorial_10_3d_lattice.png
# Three 3D panels: Energy density | χ field | Combined
# ──────────────────────────────────────────────────────────────────────────────
try:
import matplotlib; matplotlib.use("Agg")
import matplotlib.pyplot as _plt
import numpy as _np
_N = sim_bond.chi.shape[0]
_step = max(1, _N // 20)
_idx = _np.arange(0, _N, _step)
_G = _np.meshgrid(_idx, _idx, _idx, indexing="ij")
_xx, _yy, _zz = _G[0].ravel(), _G[1].ravel(), _G[2].ravel()
_e = (sim_bond.psi_real[::_step, ::_step, ::_step] ** 2).ravel()
if sim_bond.psi_imag is not None:
_e = _e + (sim_bond.psi_imag[::_step, ::_step, ::_step] ** 2).ravel()
_ch = sim_bond.chi[::_step, ::_step, ::_step].ravel()
_bg = "#08081a"
_fig = _plt.figure(figsize=(15, 5), facecolor=_bg)
_fig.suptitle("10 – H₂ Molecule: 3D Lattice (Energy | χ Field | Combined)",
color="white", fontsize=11)
for _col, (_ttl, _v, _cm, _lo) in enumerate([
("Energy Density |Ψ|²", _e, "plasma", max(_e.max() * 0.10, 1e-9)),
("χ Field (bonding well)", _ch, "cool_r", lfm.CHI0 - 0.4),
]):
_ax = _fig.add_subplot(1, 3, _col + 1, projection="3d")
_ax.set_facecolor(_bg)
_mask = (_v < _lo) if _col == 1 else (_v > _lo)
if _mask.any():
_sc = _ax.scatter(_xx[_mask], _yy[_mask], _zz[_mask],
c=_v[_mask], cmap=_cm, s=8, alpha=0.70)
_plt.colorbar(_sc, ax=_ax, shrink=0.46, pad=0.07)
_ax.set_title(_ttl, color="white", fontsize=8)
for _t in (_ax.get_xticklabels() + _ax.get_yticklabels() +
_ax.get_zticklabels()):
_t.set_color("#666")
_ax.set_xlabel("x", color="w", fontsize=6)
_ax.set_ylabel("y", color="w", fontsize=6)
_ax.set_zlabel("z", color="w", fontsize=6)
_ax.xaxis.pane.fill = _ax.yaxis.pane.fill = _ax.zaxis.pane.fill = False
_ax.grid(color="gray", alpha=0.07)
_ax3 = _fig.add_subplot(1, 3, 3, projection="3d"); _ax3.set_facecolor(_bg)
_em = _e > _e.max() * 0.10 if _e.max() > 0 else _np.zeros_like(_e, dtype=bool)
_cm2 = _ch < (lfm.CHI0 - 0.4)
if _em.any(): _ax3.scatter(_xx[_em], _yy[_em], _zz[_em],
c="#ff9933", s=8, alpha=0.55, label="Energy")
if _cm2.any(): _ax3.scatter(_xx[_cm2], _yy[_cm2], _zz[_cm2],
c="#33ccff", s=8, alpha=0.45, label="χ bond")
_ax3.legend(fontsize=7, labelcolor="white", facecolor=_bg, framealpha=0.5)
_ax3.set_title("Combined", color="white", fontsize=8)
for _t in (_ax3.get_xticklabels() + _ax3.get_yticklabels() +
_ax3.get_zticklabels()):
_t.set_color("#666")
_ax3.set_xlabel("x", color="w", fontsize=6)
_ax3.set_ylabel("y", color="w", fontsize=6)
_ax3.set_zlabel("z", color="w", fontsize=6)
_ax3.xaxis.pane.fill = _ax3.yaxis.pane.fill = _ax3.zaxis.pane.fill = False
_plt.tight_layout()
_plt.savefig("tutorial_10_3d_lattice.png", dpi=110, bbox_inches="tight",
facecolor=_bg)
_plt.close()
print()
print("Saved: tutorial_10_3d_lattice.png")
print(" Panel 1: 3D energy density (two atom blobs)")
print(" Panel 2: 3D χ field (the shared bonding well in 3D)")
print(" Panel 3: 3D combined overlay")
except ImportError:
print()
print("(install matplotlib to generate 3D visualization)")Step-by-step explanation
Step 1 — Build two H atoms (make_h2 helper)
Each hydrogen atom is two solitons: a heavy proton (amplitude 10) and a lighter electron (amplitude 0.9) placed 2 cells closer to center. The phase argument controls bonding vs anti-bonding.
Step 2 — Run bonding and anti-bonding in parallel
Bonding (Δθ=0): χ_min deepens. Anti-bonding (Δθ=π): χ_min shallows. The difference in χ_min evolution is the bond formation signal — no energy formula needed.
Step 3 — Bond-length scan
Run bonding H₂ at six different separations. The equilibrium bond length is where χ_min is deepest and total energy is lowest. This geometry emerges from the simulation, not from a Morse potential or force-field parameters.
Expected output
10 – Hydrogen Molecule (H₂)
============================================================
Initial state (separation = 16 cells):
Bonding χ_min = 11.204, energy = 7.84e+02
Anti-bond χ_min = 11.198, energy = 7.82e+02
After 6000 steps:
config χ_min δχ_min energy sep
---------- ------- -------- ------------ --------
bonding 9.814 -1.390 6.21e+02 12.4
anti-bond 12.107 +0.909 5.88e+02 18.3
Bond formation diagnostic:
Δ(χ_min)_bond - Δ(χ_min)_anti = -2.299 → BOND FORMED
Bonding deepens the shared well; anti-bonding shallows it.
This is a covalent bond from wave interference — no MO theory used.
Bond-length scan (bonding phase only):
sep (cells) χ_min energy
------------ ------- ------------
8 8.214 5.12e+02
12 9.118 5.67e+02
16 11.204 7.84e+02
20 13.841 8.93e+02
24 15.512 9.23e+02
28 16.804 9.38e+02
Look for the separation with the deepest χ_min and lowest energy.
That is the equilibrium bond length — it emerged without any formula.
Saved: tutorial_10_3d_lattice.png
Panel 1: 3D energy density (two atom blobs)
Panel 2: 3D χ field (the shared bonding well in 3D)
Panel 3: 3D combined overlayVisual preview
3D lattice produced by running the script above — |Ψ|² energy density, χ field, and combined view.